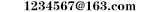肝豆状核变性发病原因和机制
(资料来源于网络,特致著作权人感谢)
发病原因
肝豆状核变性为常染色体隐性遗传性疾病。绝大多数限于同胞一代发病或隔代遗传,罕见连续两代发病。致病基因ATP7B定位于染色体13q14.3,编码一种个氨基酸组成的铜转运P型ATP酶。ATP7B基因突变导致ATP酶功能减弱或消失,引致血清铜蓝蛋白(ceruloplasmin,CP)合成减少以及胆道排铜障碍,蓄积在体内的铜离子在肝、脑、肾、角膜等处沉积,引起进行性加重的肝硬化、锥体外系症状、精神症状、肾损害及角膜色素环(Kayser-Fleischerring,K.F环)等。ATP7B基因的变异位点繁多,人类基因组是数据库中记载达多个位点。基因突变位点具有种族特异性,因此基因检测位点的选择要有针对性。我国WD患者的ATP7B基因有3个突变热点,即RL,PL和TM,占所有突变的60%左右。近年来有研究发现除ATP7B以外其他基因如COMMD1,XIAP,Atox1等也与该病相关。
发病机制
正常人每日自肠道摄取少量的铜,铜在血中先与白蛋白疏松结合,在肝细胞中铜与α2-球蛋白牢固结合成具有氧化酶活性的铜蓝蛋白。循环中90%的铜与铜蓝蛋白结合,铜作为辅基参与多种重要生物酶的合成。铜在各脏器中形成各种特异的铜-蛋白组合体,剩余的铜通过胆汁、尿和汗液排出。
疾病状态时,血清中过多的游离铜大量沉积于肝脏内,造成小叶性肝硬化。当肝细胞溶酶体无法容纳时,铜即通过血液向各个器官散布和沉积。基底节的神经元和其正常酶的转运对无机铜的毒性特别敏感,大脑皮质和小脑齿状核对铜的沉积也产生症状。铜对肾脏近端小管的损害可引起氨基酸、蛋白以及钙和磷酸盐的丢失。铜在眼角膜弹力层的沉积产生K-F环。与此同时,肝硬化可产生门静脉高压的一系列变化。
病理生理
病理改变主要累及肝脑肾角膜等。肝脏表面和切片均可见大小不等的结节或假小叶,逐渐发展为肝硬化,肝小叶由于铜沉积而呈棕黄色。脑的损害以壳核最明显,苍白球、尾状核、大脑皮质、小脑齿状核也可受累,显示软化、萎缩、色素沉着甚至腔洞形成。光镜下可见神经元脱失和星形胶质细胞增生。角膜边缘后弹力层及内皮细胞浆内有棕黄色的细小铜颗粒沉积。
铜娃娃罕见病关爱中心中国肝豆状核变性罕见病关爱协会/铜娃娃罕见病关爱中心,ChineseOrganizationforWilsonDisease,简称COWD,Wilson-China,是由肝豆状核变性罕见病患者及其家属、志愿者等自发组织的民间非营利性公益机构,是中国目前唯一一家专注于Wilson病群体关爱服务的病友组织。Wilson-China,中国肝豆状核变性(Wilson病)病友和家属交流分享战“豆”经验的精神家园,传播wilson病的正确知识和治疗进步,自助,助人,直面疾病,共同携手推动肝豆罕见病的救助和政策进步,为罕见病患者争取更多的尊重及平等的生存机会、生活质量!
同病相连,不“铜”人生!
联系我们


 当前时间:
当前时间:  E-Mail:
E-Mail: